
Research Program
The ‘Interfacing image analysis and molecular life science’ (Imol) research group will educate PhD students in the theory and practice of advanced microscopy methods, cutting-edge image analysis and apply these technologies on neuroscience applications. Microscopy is instrumental to our understanding of the basic principles of life at multiple spatio-temporal scales and resolutions (Figure 1). Recent technological developments in light and electron microscopy have broadened and deepened our understanding of biological processes. The Imol reseach group emphasizes on neuroscience application such as
(i)the analysis of cell-cell junctions between epithelial cells that affect the blood-brain barrier,
(ii)how vascular and neuronal structures intermingle along the developmental process in order to generate functional organs and
(iii)at neurodegeneration and the effect of the formation of extracellular amyloid plaques.
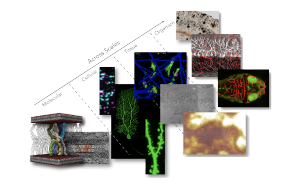
Figure: Imaging neuroscience applications across scales. From the molecular, cellular, tissue and organism level a multitude of informations collected in order to address various questions essential to life.
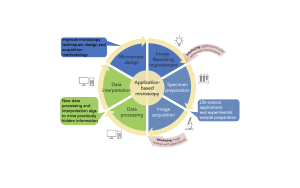 Figure: The cyclic approach of Imol RTG provides distinct entry points (red arrows) that allow us to interface currently decoupled research areas. Yellow arrows indicate traditional microscope design and hypothesis development as well as specimen preparation and image acquisition. Subsequent data processing and data interpretation leads to results. Red arrows indicate entry points for algorithm developers, hardware developers and biologists working at the interfaces of software/hardware, microscopy/biology, biology/data mining. We expect these interfaces to lead to significant improvements in all areas of microscopy.
Figure: The cyclic approach of Imol RTG provides distinct entry points (red arrows) that allow us to interface currently decoupled research areas. Yellow arrows indicate traditional microscope design and hypothesis development as well as specimen preparation and image acquisition. Subsequent data processing and data interpretation leads to results. Red arrows indicate entry points for algorithm developers, hardware developers and biologists working at the interfaces of software/hardware, microscopy/biology, biology/data mining. We expect these interfaces to lead to significant improvements in all areas of microscopy.
To do this we will
(i)improve microscopy techniques from a technological perspective, in terms of both conceptual design and acquisition methodology
(ii)Address the life-science applications and experimental sample preparation conditions that facilitate the recording of the highest-quality images.
(iii)Develop new data processing and interpretation algorithms that can mine previously inaccessible information.